


Introduction
Isochromosomes are supernumerary chromosomes that are made up of two copies of the same arm on a chromosome [1-3]. Tetrasomy 18p was first reported by Froland et al. in 1963 [4]. It is a very rare chromosomal anomaly with a prevalence of 1/140,000-180,000 but is also one of the most commonly observed isochromosomes, and affects both genders equally [1-4]. Tetrasomy 18p syndrome is characterized by nonspecific morphologic features; low birth weight, microcephaly, low-set ears, strabismus, abnormalities in muscle tone and deep tendon reflex [1,3,5,6]. Feeding difficulties and developmental retardation are also followed [2,6]. Cardiac and renal malformations are rare, therefore, mortality rate is low [1,5,7]. Because of its very low prevalence rate, tetrasomy 18p has not yet been reported in Korea. Herein, we report the first case of prenatally diagnosed tetrasomy 18p.
Case report
A 28-year-old primi gravid woman was referred to our fetal treatment center because of suspected fetal congenital heart disease at 32+4 weeks of gestation. The ultrasonography showed asymmetric intrauterine growth retardation (IUGR) with 5-week smaller abdominal circumference. The fetal echocardiography demonstrated dextrocardia with cardiomegaly (cardio-thoracic ratio, 0.64), mild pericardial effusion, and decreased left ventricular function (modified myocardial performance index, 0.68). There was no intracardiac abnormality. Doppler findings in the middle cerebral artery revealed increased peak systolic velocity (71 cm/sec, 1.3-1.5 MoM), which suggested fetal anemia. Imperforate anus was also suspected. The cordocentesis was performed at 33+2 weeks of gestation for identifying the karyotype, hemoglobin, and the presence of viral infection. The karyotyping confirmed tetrasomy 18p by G-banding and fluorescence in situ hybridization (Fig. 1). Fetal hemoglobin level was 9.7 g/dL and there was no evidence of viral infection such as toxoplasma, rubella, cytomegalovirus, and herpes simplex virus.
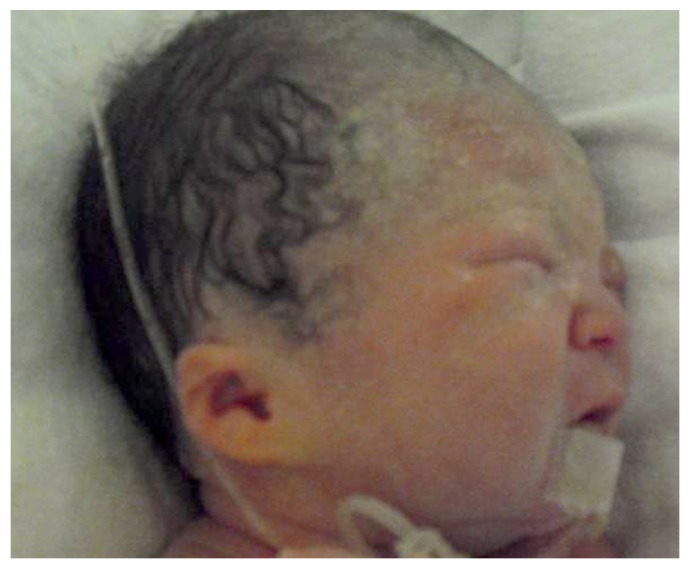
The male infant was delivered at 36+6 weeks of gestation; weighing 2,256 g, which was below 10 percentile. Apgar score was 6, 8 at 1, 5 minutes, respectively. Because of low oxygen saturation (76%), the baby was admitted to the neonatal intensive care unit. Initial hemoglobin level was 11.4 g/dL, and after transfusion of packed red blood cells, his oxygen saturation got over 97% without applying oxygen. Low-set ears with small auricles were shown (Fig. 2) and muscle tone was increased. The postnatal echocardiography and cardiac computerized tomography revealed dextrocardia and mild cardiomegaly without pericardial effusion, and the cardiac function was within normal range. As prenatally suspected, the baby had a low type of imperforate anus, and underwent anoplasty. Swallowing difficulty was also found and after feeding rehabilitation therapy, the baby could be fed. There were no other abnormalities on further evaluations.
At the age of 3 months, the brainstem auditory evoked potentials and auditory brainstem response revealed right sensorineural hearing loss due to the peripheral conduction defect. The follow-up echocardiography still showed dextrocardia without cardiomegaly. The baby was scheduled for the developmental test.
Discussion
Tetrasomy 18p is a duplication of the short arm and deletion of the long arm on chromosome 18 [3,5]. It is a very rare chromosomal abnormality with a prevalence of one in 140,000 to 180,000 live births, affecting males and females equally [2,3,7]. Most cases are reported to be de novo formation from parents with normal karyotypes, however, there are also some case of mosaic type [1,3,4,6-8]. In the present report, the karyotypes of the parents were normal, indicating that the affected baby also had a de novo formation. Increased paternal or maternal age is considered to be relevant to tetrasomy 18p because of higher rates of centromeric nondisjunction or misdivision of chromosome 18 during the second phase of meiosis [3,4,9-11].
Tetrasomy 18p demonstrates various characteristic features; low birth weight, microcephaly, low-set ears, short palpebral fissures, high nasal bridge, abnormal muscle tone, feeding difficulties, developmental delay, and mental retardation [2,8]. There could be occurrence of strabismus, recurrent otitis media, cryptorchidism, scoliosis/kyphosis [2-5,8,9]. In the present report, the fetus showed IUGR, dextrocardia with cardiomegaly, anemia and imperforate anus. The baby also had low birth weight, low-set ears, feeding difficulties and increased muscle tone. Because of its variety of clinical features and its rarity, it is difficult to diagnose tetrasomy 18p prenatally.
Combined cardiac anomaly has been reported to be rare in tetrasomy 18p. According to the review of tetrasomy 18p by Sebold et al. [2], which reported most cases with cardiac anomaly, 12.3% of tetrasomy 18p patients had cardiac anomalies including patent foramen ovale, ventricular or atrial septal defects, and some of them also showed cardiac dysfunction, such as valvular regurgitation or stenosis. However, the present fetus did not have such cardiac anomaly or dysfunction, except for the dextrocardia. Although cardiomegaly with mild pericardial effusion was found prenatally, it was resolved spontaneously after transfusion. Fetal anemia can cause volume overload and redistribution of body fluid, leading to fetal cardiomegaly with pericardial effusion, ascites or hydrops [12]. Postnatal laboratory test identified that fetal anemia was due to iron deficiency from postnatal laboratory tests. Even there is no report about fetal anemia in tetrasomy 18p, based on the present case, fetal anemia could be associated with tetrasomy 18p.
As shown in the present report, when the fetus showed IUGR, anemia and combined facial dysmorphism, we recommend performing karyotyping and study for viral infection. After making a diagnosis chromosomal anomaly, it is crucial to refer the patient to the tertiary center and provide genetic counseling followed by subsequent medical and behavioral management in order to enhance the quality of life of the affected individuals and their families.







")











